
|
Manual |
The C(s) analysis [P. Schuck (2000) Size distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophysical Journal 78:1606-1619.] can be used to generate sedimentation coefficient distributions from sedimentation velocity experiments. The principle of the method is based on finding non-zero terms in a linear combination of Lamm equation solutions describing the individual components in a sample. The coefficients of each term determine the partial concentration of each component. The method requires input of an upper and lower s-value boundary, an f/f0 frictional ratio, and a sedimentation coefficient resolution setting that determines the granularity of the linear combination. The frictional ratio is used to calculate a corresponding diffusion coefficient for each s-value term in the linear combination.
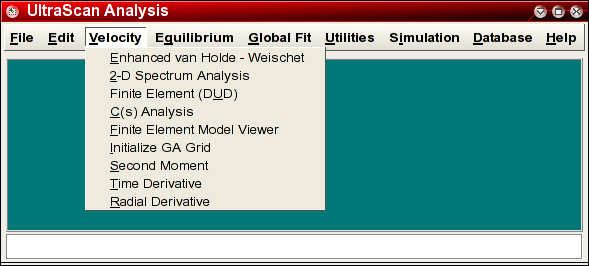
The C(s) analysis can be started in one of two ways: First, you can launch the application by clicking on "C(s) Analysis" in the Velocity sub-menu of the main menu. The main C(s) analysis window will appear. The second way to launch this application is from within the enhanced van Holde - Weischet Analysis. By using the van Holde - Weischet analysis, a model independent approach, a limit for the possible s-values present in the sample under investigation can be established before the C(s) analysis is performed. This eliminates the uncertainty introduced when the user selects these limits without prior knowledge, and it is therefore the preferred method to select the limits and to launch the application.
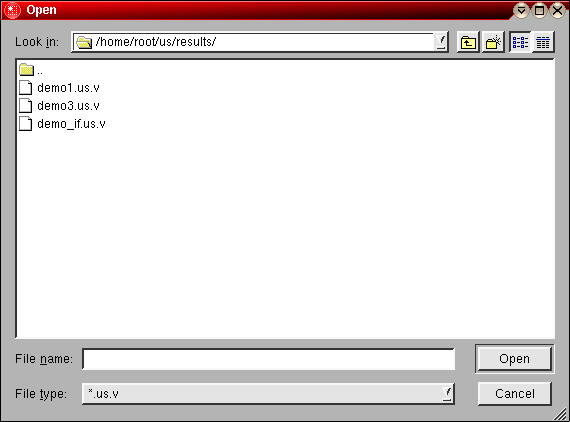
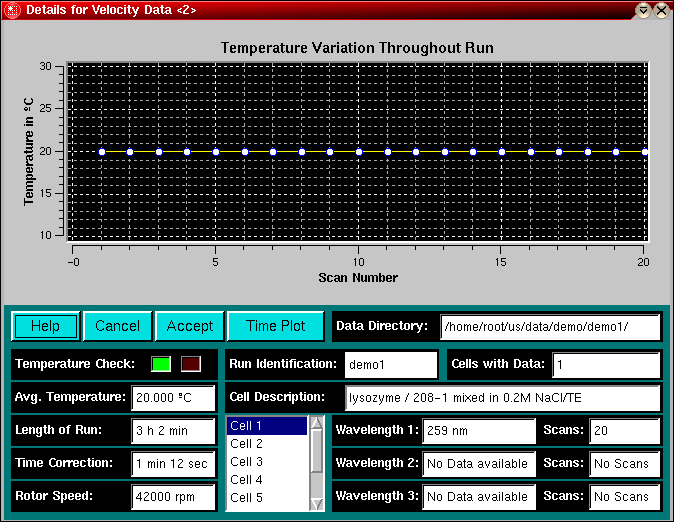
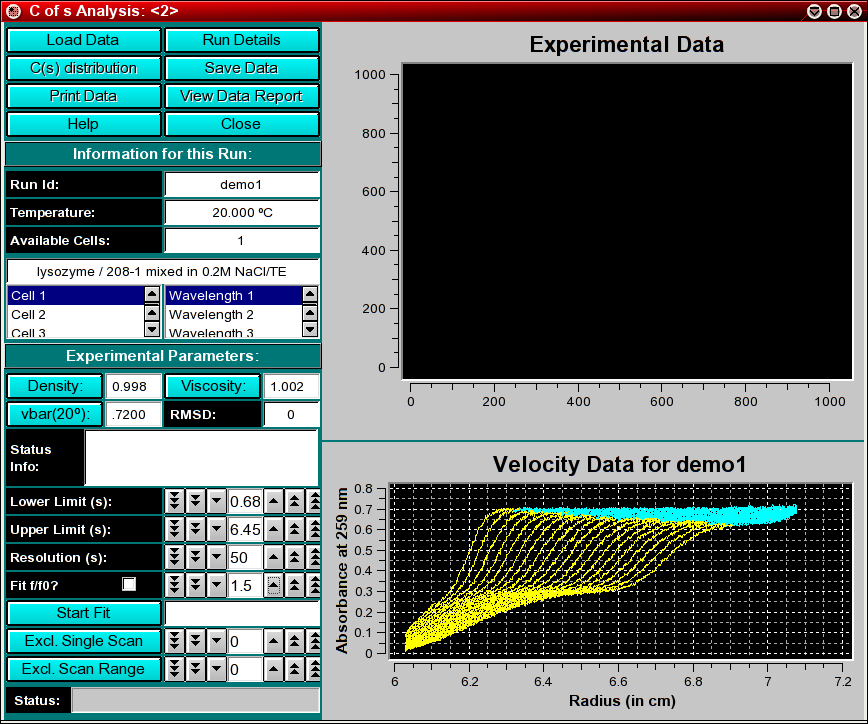
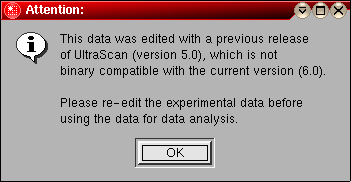
The first step in the analysis requires that you load an UltraScan dataset that has previously been edited with the UltraScan editing module. UltraScan sedimentation velocity datasets always have the suffix ".us.v". Click on the "Load Dataset" button in the left upper corner of the control panel. Simply select the desired run from the selection of available runs in the dataset loading dialog. Once loaded, the Run Details will be shown. Click on the "Accept" or "Cancel" button, and you will be returned to the analysis window, which will show the first available dataset of the selected run in the edited data window on the lower right panel.
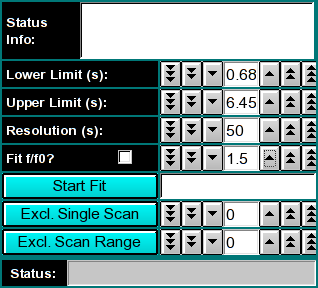
Next, confirm the upper and lower limits for the s-value distribution range to be fitted to your data. If you launched the application from the enhanced van Holde - Weischet analysis, these values will already be set to a suggested value, otherwise, a default range between 1s - 100s will be shown (which will not be appropriate for most applications). Also, a resolution setting needs to be selected. 20-50 is an appropriate default for the first analysis. This can be refined in subsequent iterations. Finally, an f/f0 value has to be selected. An f/f0 value of 1 corresponds to a molecule with a perfectly spherical shape, the larger the value is, the more non-globular the simulated shape will be. If you are not sure of the appropriate value for f/f0, leave it at the default value of 1.5.
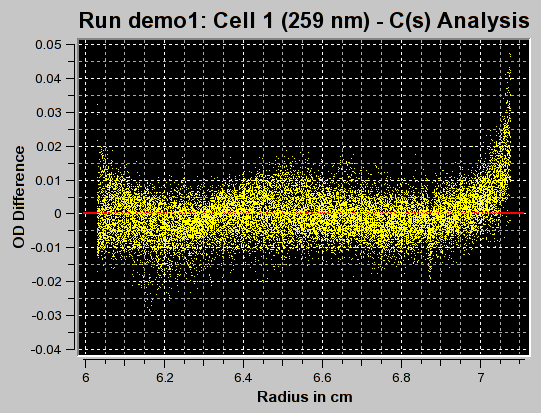
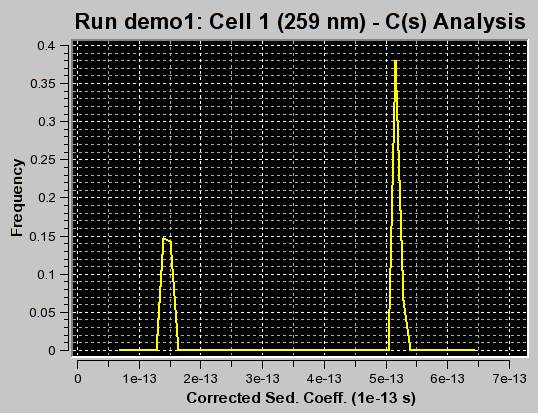
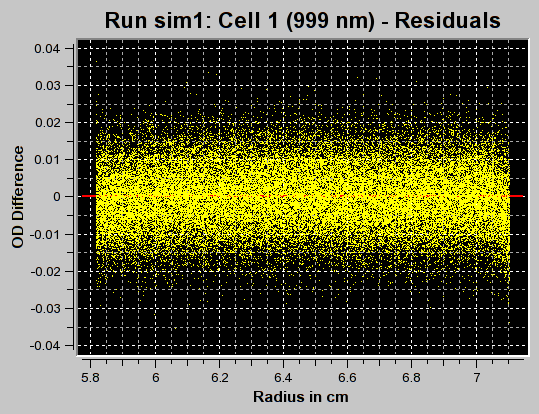
The first iteration should be made with the "Fit f/f0?" value left unchecked by clicking on the "Start Fit" button. A single iteration will be fitted (which will be fast) and provide an initial view of the s-value distribution appropriate for the sample. During fitting, the "Start Fit" button will change text to "Stop Fit". Clicking on it at any time during the fit allows you to abort a currently active fit. After the iteration is completed, the upper plot panel will show now the residuals of the fit. A red/green residuals pixels map is also shown in a separate window. Please refer to this link for more information on the residuals pixel map. The C(s)distribution can now be shown by clicking on the "C(s) Distribution" button in the upper left control panel. Once clicked on, the button will change text to show "MW Distribution". Clicking again, the fitted f/f0 value will be used to transform the C(s) distribution into a molecular weight distribution. At this point the button will change text again to show "Residuals". Repeatedly clicking this button will cycle through these three options.
Continue to adjust the upper and lower limits until an acceptable range of s-values has been determined by performing single fits. Generally, such a range is found when the spacing of peaks is well distributed throughout the range and no end effects are visible (large amplitudes at the end points of the distribution).
At this point one can fit for the most representative f/f0 value by selecting the f/f0 fitting checkbox. Clicking on the "Start Fit" button will then start a fitting session that tries to determine the optimal f/f0 value.
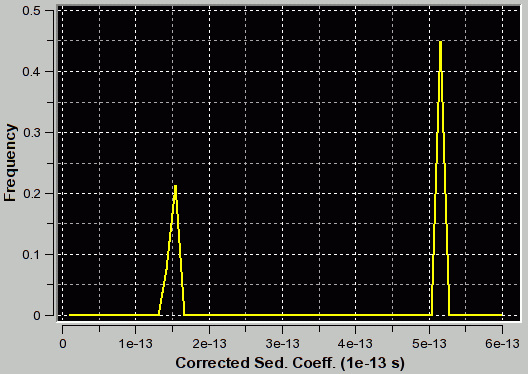 |
f/f0=1.2 (appropriate for first peak and corresponding to the true C(s) distribution as verfified by van Holde - Weischet and finite element analysis) |
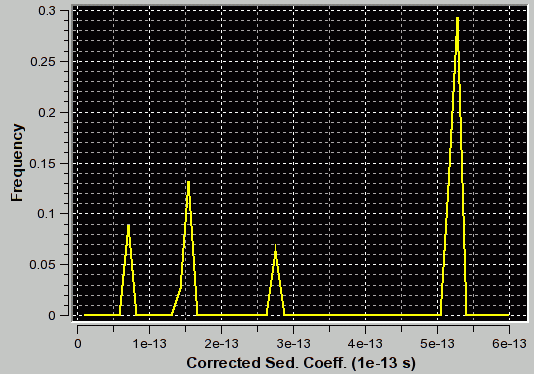 |
f/f0=3.1 (appropriate for second peak) |
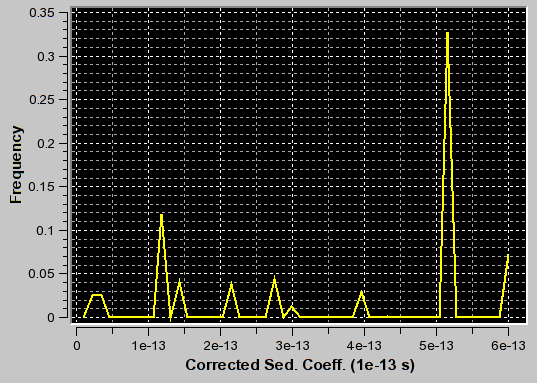 |
f/f0=3.1 (appropriate for second peak) |
 |
Click on these buttons to control the enhanced van Holde - Weischet analysis.
|
Run Information:
 |
|
Experimental Parameters:
 |
Here you can enter the corrections for density, viscosity, and partial
specific volume of your sample. As you change the information in these
fields, the program will automatically update the analysis to correct the
S-value according to the specified buffer conditions. If the data was
retrieved from the database and associated with the correct buffer file
and peptide file, the density, viscosity and partial specific volume (vbar)
will automatically be updated to the appropriate values and don't need to be
adjusted to produce S 20,W corrected values.
|
Analysis Controls:
 |
|
This document is part of the UltraScan Software Documentation
distribution.
Copyright © notice.
The latest version of this document can always be found at:
Last modified on January 27, 2005.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}