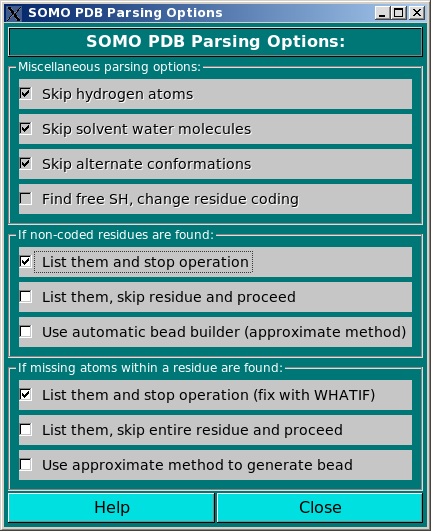
In this module, still under development, you can control the options affecting the
way SOMO reads and loads the various fields present in PDB files.
 |
|
In the Miscellaneous parsing options subpanel, the four options listed are
not presently selectable. The first three are currently hard-coded in the
program, while the fourth is in the planning stage.
-
Skip hydrogen atoms. Explicit hydrogen atoms are found in NMR- or neutron
crystallography-derived structures, and are not coded for in the distributed
somo.residue file. Therefore, the parser automatically skips any line containing
them. In the current SOMO implementation, the hydrogen atoms are "included" in the heavy
atoms to which they are bound, but we do not exclude to make the skipping as an option
to allow users to explicitly treat hydrogens; obviously, this will call for a re-definition
of all atoms and residues in the somo.hybrid, somo.atom, and somo.residue files.
-
Skip solvent water molecules. In the current SOMO implementation, the water
of hydration is treated in a statistical manner. Therefore, lines in PDB files coding for
explicit crystallographic water molecules are automatically skipped. However, we plan to make
this as an option in the future, since one might wish to investigate the effect of the
crystallographic waters on the hydrodynamics, or to use artificial hydration schemes
employing explicit water molecules. As for the previous field, using explicit waters will
also call for a re-definition of the hydration numbers in the somo.residue file.
-
Skip alternate conformations. PDB files sometimes contain alternate coordinates
for particular atoms within residues. It is presently not possible to explore the effect
of these alternate conformations, which should be negligible anyway. Therefore, presently
SOMO uses only the first ("A") alternate conformation by default. In the future, we might
provide the choice between the alternate conformations.
-
Find free SH, change residue coding. In the somo.residue file, cysteine
(CYS) is treated as if it's always engaged in a S-S bond with another cysteine (cystine). This
is because currently there's no distinction in PDB files between cysteines and cystines. Since
this involves slightly different physico-chemical parameters (e.g., two H atoms are lost when
a S-S bridge is formed), we plan to offer an automatic "free SH" finder by checking the distances
between all sulphur atoms in CYS residues. Those that won't be within bonding distance from another CYS will be considered as free SH, and their residue name changed to CYH, for which the coding is already provided within the somo.residue file. Users wishing to deal with this problem now
can manually edit their PDB file and change to CYH the name of the CYS residues that they know are not
S-S bonded.
|
The next two subpanels deal with non-coded residues and missing atoms, respectively.
 |
|
In the If non-coded residues are found: subpanel, the user will be able to choose between three
options:
-
List them and stop operation. This is the most rigorous option, and calls for
properly define the "new" residue in the somo.residue (and in the somo.atom, if
"new" atoms are also present) file(s). The program is therefore halted, waiting for proper action
to be taken. Presently, this is the default option for this subpanel.
-
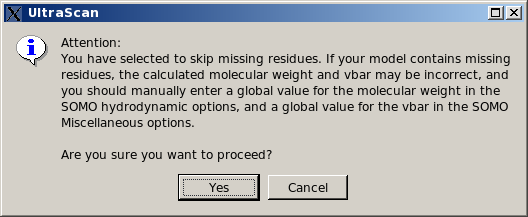
List them, skip residue and proceed. This is the least rigorous option, as skipping
residues will affect the computations on two levels. First, if the skipped residues are exposed,
their contribution to the hydrodynamics will be missed. Second, their contribution to the molecular
weight and partial specific volume of the structure will be also missed, although this can be
bypassed by entering global values in the appropriate fields (respectively in the
SOMO Hydrodynamic Calculation Options and
Miscellaneous Options panels). Therefore, this option should be used only if the skipped residue(s) is (are) known to not likely contribute to the hydrodynamics, and by entering appropriate global values for the molecular weight and partial specific volume. Be aware, however, that the total volume of the bead model, used in the Volume Correction, will likely be underestimated (unless the skipped residues are all buried), thus affecting the computation of the Rotational Diffusion Coefficient and of the Intrinsic Viscosity (see here for more explanations on this subject).
In any case, if this option is selected, the pop-up window shown below will appear warning of the potential errors and suggesting to enter appropriate values for the global molecular weight and partial specific volume.
|
 |
-
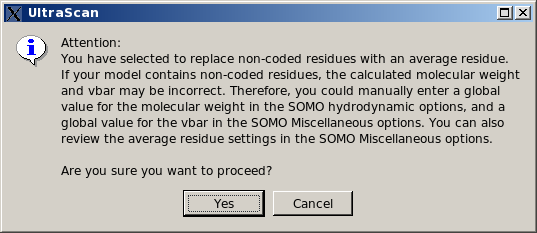
Use automatic bead builder (approximate method).
This is a "patch" option, allowing to roughly define a single "side-chain" bead for each non-coded residue. This procedure is based on an "average" volume for each atom (with an "average" molecular weight and hydration number), from which a global volume (and molecular weight) is calculated. An "average" radius for each atom is
also provided for the ASA routines (see here). The bead is then placed at the center of mass of all the atoms within the non-coded residue, and an "average" partial specific volume and color-coding ("10", light green) are also assigned to it. All these "average" values can be modified in the
Miscellaneous Options panel. However, the program will first
perform a check to recognize if the non-coded residue has the structure of an amino acid (i.e., if the N, CA, C, and O atoms are
all present). In this case, it will generate a standard "peptide bond" bead for these atoms, and a second bead for the non-coded side chain using the approximate method. In any case, as with the previous option, the approximations introduced in the molecular weight and partial specific volume can be bypassed by entering global values in the appropriate fields (respectively in the SOMO Hydrodynamic Calculation Options and in the
Miscellaneous Options panels. As for the previous option, if this option is selected a pop-up window will appear asking for entering appropriate values for the global molecular weight and partial specific volume, and prompting to check the approximate method parameters.
|
 |
|
 |
|
In the If missing atoms within a residue are found: subpanel, the user will be able to choose between three
options:
-
List them and stop operation (fix it with WHATIF). This is the most rigorous option, and calls for either manually build the missing atoms in the PDB file, or, for proteins, to use an automatic missing atom(s) builder such as WHATIF (http://swift.cmbi.ru.nl/servers/html/index.html; WARNING: WHATIF will not build missing main chain atoms, only
side chain atoms). The program is therefore halted, waiting for proper action to be taken. Presently, this is the default option for this subpanel.
-
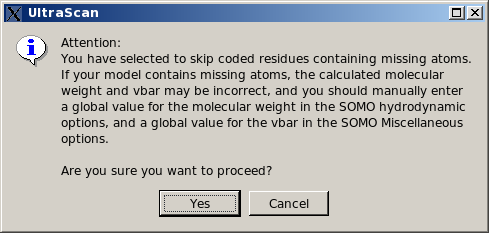
List them, skip entire residue and proceed. This is the least rigorous option, as skipping residues will affect the computations on two levels. First, if the skipped residues are exposed, their contribution to the hydrodynamics will be missed. Second, their contribution to the molecular weight and partial specific volume of the structure will be also missed, although this can be bypassed by entering global values in the appropriate fields (respectively in the SOMO Hydrodynamic Calculation Options and
Miscellaneous Options panels). Therefore, this option should be used only if the skipped residue(s) is (are) known to not likely contribute to the hydrodynamics, and by entering appropriate global values for the molecular weight and partial specific volume. Be aware, however, that the total volume of the bead model, used in the Volume Correction, will then likely be underestimated (unless the skipped residues are all buried), thus affecting the computation of the Rotational Diffusion Coefficient and of the Intrinsic Viscosity (see here for more explanations on this subject).
In any case, if this option is selected, the pop-up window shown below will appear suggesting to enter appropriate values for the global molecular weight and partial specific volume. The reason why only the missing atoms cannot be skipped is because they might be used to define the position of the bead, so their absence cannot be tolerated.
|
 |
-
Use approximate method to generate bead.
This is a "patch" option, allowing to keep the original bead definitions. It is based on the assumption that the missing atoms were present in the original macromolecule, but could not be located experimentally. If the missing atoms are not among those defining the position of the bead, nothing is really affected. When one or more (but not all) the atoms determining the position are missing, the remaining ones are used to position the bead. If all the position-determining atoms are missing, then the bead will be positioned at the cog of all the atoms present. If all the atoms of a bead are missing, their parameters
(mass, volume, hydration) will be added to those of the next bead, creating a single bead positioned on the cog of all atoms present. For instance, for an amino acid completely missing the side-chain, the peptide bond size and mass will be that of the original peptide bond plus that of the missing side chain, positioned as a regular peptide bond bead. For amino acids, if the missing atoms
belong to the peptide bond segment, then the peptide bond rule (see here) will be disallowed.
If coded residues contains extra or non-coded atoms, they will be treated as non-coded residues with the automatic bead builder. As for the previous option, if this option is selected a pop-up window will appear asking if it's OK to proceed.
|
 |